Epithelial–mesenchymal interactions (EMIs) are critical for tooth development. Molecular mechanisms mediating these interactions in root formation is not well understood. Laser capture microdissection (LCM) and subsequent microarray analyses enable large scale in situ molecular and cellular studies of root formation but to date have been hindered by technical challenges of gaining intact histological sections of non-decalcified mineralized teeth or jaws with well-preserved RNA. Here, we describe a new method to overcome this obstacle that permits LCMof dental epithelia, adjacent mesenchyme, odontoblasts and cementoblasts frommouse incisors and molars during root development. Using this method, we obtained RNA samples of high quality and successfully performed microarray analyses.
Robust differences in gene expression, as well as genes not previously associated with root formation, were identified. Comparison of gene expression data from microarray with real-time reverse transcriptase polymerase chain reaction (RT-PCR) supported our findings.These genes include known markers of dental epithelia, mesenchyme, cementoblasts and odontoblasts, as well as novel genes such as those in the fibulin family. In conclusion, our new approach in tissue preparation enables LCM collection of intact cells with well-preserved RNA allowing subsequent gene expression analyses using microarray and RT-PCR to define key regulators of tooth root development.International Journal of Oral Science (2012) 4, 7–13; doi:10.1038/ijos.2012.15; published online 16 March 2012
Keywords:dental laser tips; laser capture microdissection;root;dental laser handpiece
INTRODUCTION
Tooth formation proceeds through a series of highly orchestrated reciprocal signaling interactions between dental epithelial and mesenchymal cells, termed epithelial–mesenchymal interactions (EMIs).1–5 Many genes/proteins involved in this process were identified and mapped temporally and spatially.6–7In contrast to the crown formation, very few regulators of the root formation were identified.8–11 Work by our group and others suggests a role of epithelial signals in inducing the root development. The root forming cells such as cementoblasts (CB), dental follicle (DF) cells and other dental mesenchymal cells respond to enamel matrix proteins both in vitro and in vivo.11–18 Further support for a role of epithelial products in root development comes from tooth organ culture data and from investigation of genes expressed by Hertwig’s epithelial root sheath.17,19–20 The data from these studies suggest that genes expressed by epithelial cells during root development are significantly different from those expressed during early stages of crown development; however, in depth studies to confirm these proposals, as well as knowledge as to specific similarities and/or differences in genes/proteins for crown vs. root development are minimal.
To determine the role of specific genes during tooth development,gene knockout animal models are often used. Many of these animalmodels are lethal and thus are only suitable for examining early stages of tooth development, i.e., crown development, since in rodents,first molar crown development is almost complete prior to birth.
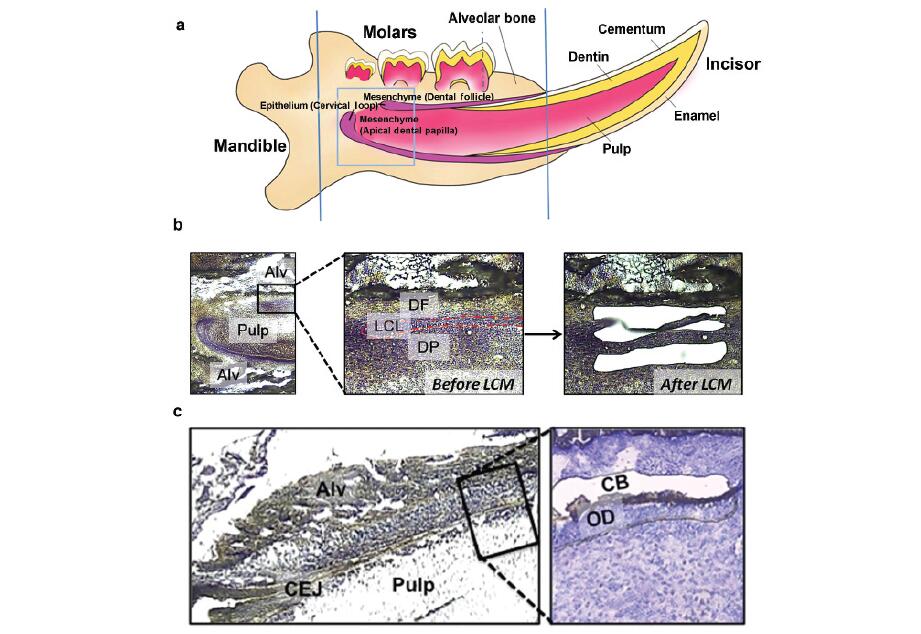
By limiting gene ablation to specific tissues and thus allowing for longer lifespans, conditional gene knockout models have served some purposes; however, without having specific markers for targeting tooth root development, such models have limited value.21–22 Laser capture microdissection (LCM) has been used extensively to analyze gene expression profiles in various non-mineralized tissues 23–24 and in the developing teeth at early stages of crown formation when the tooth is not highly mineralized.25–29 In mineralized tissue research, the difficulty in preparing intact unfixed, undecalcified and highlymineralized tissue sections (such as dentin and enamel)withwellpreserved cellular structures and high-quality RNA for LCM is a common obstacle.Over the past several years,we have optimized a technique for LCM of cells associated with the dento-alveolar bone complex. In this study, we aim to describe a new approach for tissue preparation and cell collection using LCM of postnatal day 7 (P7) and P14 mouse mandibles. This technique facilitates the use of LCM and microarrays to delineate regulatory mechanisms of the root development.
Mouse incisors are continuously erupting and have all stages of differentiating cells, while their molars develop in a similar fashion to humans. Therefore, we used both mouse incisors and molars in this study to define the regulatory mechanisms associated with the root formation and determine whether the EMIs are critical for the development of root tissues.30–32 This information should provide valuable insights and thus inform periodontal tissue regenerative therapy and tooth (crown and root) regeneration in the future.
MATERIALS AND METHODS
Sample preparation As shown in Figure 1, lingual cervical loop (LCL) epithelial cells, adjacent apical dental papilla (DP) and apical DF cells were collected from mandibular incisors of threeCD-1 mice on P7.Odontoblasts(OD) and CB were collected from roots of mandibular first molars from three mice on P14when molar roots are actively developing. The protocol for animal use was approved by the University ofWashington Institutional Animal Care and Use Committee. All equipment used for surgical dissectionbof mandibles was cleaned with RNaseZap (Applied Biosystem/Ambion, Austin, TX, USA). After the erupted portion of mandibular incisors and excess bones were cut off (Figure 1a), the mandibles were embedded in an optimal cutting temperature (Tissue-Tek; Sakura Finetek USA, Torrance, CA, USA) block using standard cryomolds (Sakura Finetek USA) and stored at 280 6C until use.For P7 incisor cervical loop epithelial and adjacent mesenchymal samples, the frozen mandibles were sectioned sagittally at a thickness of 14 mmin a cryostat (CM3050 S; Leica Microsystems, Buffalo Grove,IL, USA) using a disposable tungsten carbide blade (TC-65; Leica Microsystems, Buffalo Grove, IL, USA). For P14 root OD and CB,the frozen mandibles were sectioned frontally at a thickness of 6 mm at 220 6C. Six sections were placed per polyethylene naphthalate membrane frame slide (MDS Analytical Technologies, Sunnyvale,CA, USA) and kept at 280 6C until staining and the laser capture microdissection were performed.
Gene expression microarray analysis Fifty nanograms of each total RNA sample were used for RNA amplification using Ovation Pico WTA System (NuGEN Technologies,San Carlos, CA, USA). Each amplified antisense cDNA product was converted into a sense cDNA transcript (ST-cDNA) and fragmented using WT-Ovation Exon Module (NuGEN Technologies). Each fragmented cDNA product was biotinylated, using an Encore Biotin Module according to the manufacturer’s instructions (NuGEN Technologies).
Each biotinylated probe was hybridized onto a GeneChip Mouse Gene 1.0 ST Array (Affymetrix, Santa Clara, CA, USA) for 16 h at 456C. After hybridization, the array chips were washed, stained with streptavidin phycoerythrin using an Affymetrix Fluidics Station 450 and imaged using an Affymetrix Gene Chip Scanner 3000(Affymetrix).
RESULTS
RNA integrity Using the protocol described in the ‘Materials and methods’ section, we were able to obtain a sufficient quality and quantity ofRNAsamples for global gene expression analyses using whole genome expression microarrays (Affymetrix mouse gene 1.0 ST). The RIN of each sample is shown in Table 1. RINs of all RNA samples were above or close to 6.
Instead of using traditional in vitro transcription from cDNA carrying the T7 promoter, we used Ribo-SPIA reactions with a combination of universal RNA primer, DNA polymerase and RNAse to isothermally amplify total RNA (Ovation Pico WTA System; NuGEN Technologies).Using this technique, we found that RNA samples with RIN above or close to 6 can be obtained and used successfully for whole genome microarray (Affymetrix mouse gene 1.0 ST).
Quality of gene expression data from microarrays Data for quality control metrics of each array are shown in Table 2.Every array passed all quality control metrics. The area under the curve value for a receiver operating characteristic (ROC) curve plotted detection of positive controls against false detection of negative controls(Pos_vs_neg_auc) ranged from 0.84 to 0.89 for all arrays, indicating good array hybridization and signal quality for each gene expression microarray chip. Arrays were neither dim nor denoted as an outlier (Table 2 and Figure 2). After RMA expression, values were computed, and background correction and relatively identical distribution of expression values across arrays were ensured, indicating good array data quality.



